Drug-Device-Combinations
General Background
Drug-Device-Combinations (DDCs) represent a heterogenous group of products, which are a combination of medical devices on one hand and pharmaceutical products on the other. This product group has evolved significantly over the past years as it bears a high potential of clinical benefits for treatment of various types of diseases. Among others, DDCs may benefit patients with regular and long-term dosing requirements in an outpatient setting, either by self-administration or with the support of a professional user. This reduces the burden on patients and on healthcare systems, supporting compliance and safety of medicinal uptake by patients.
The product spectrum ranges from simple prefilled syringes (filled with a drug component), auto injector devices to complex “smart” drug-delivery on demand systems which are implanted (e.g. insulin pumps). Other devices represent a combination of a traditional medical device (e.g. a stent) coated with a pharmaceutical to improve the clinical outcome of the treatment (e.g. drug eluting stent).
Nomenclature and applicable legislation
DDC’s always combine a medical device with a medicinal product leading to applicability of two different legislations. In Europe, those legislations are the EU Medical Device Regulation (Regulation 2017/745) on one hand and the Medical Products Directive (Directive 2001/83/EC) on the other.
The nomenclature is not yet used consistently in Europe; Basically, it is often differentiated between integral and non-integral DDC products.
Non-integral DDC products: There are cases where a medicinal product and a medical device are placed on the market in the same secondary packaging (= co-packed) but do not form an integral product, for example, a vial containing a medicinal product solution, with an (empty) CE marked sterile syringe. This product is NOT considered a drug-device-combination as the medical device falls under the first subparagraph of Article 1(9) of the EU-MDR.
Furthermore, devices referenced in the medicinal product information, or medicinal products referenced in the information supplied with the device, are NOT considered-drug-device-combinations. Requirements for these products are covered in section 3 of the Q&A of the guidance EMA/37991/2019. If in doubt, whether a specific device is a DDC, it is suggestive to consult the MDCG 2022-5: Guidance on Borderline.
Irrespective of the nomenclature, the applicable “leading” legislation is always unequivocal and depends on the principle mode of action of the product. Provided the principle mode of action is provided by the medical device part, leading legislation is the EU-MDR. In the other case, the Medical Products Directive (MPD = Directive 2001/83/EC) is the primary applicable legislation (see the figure below).
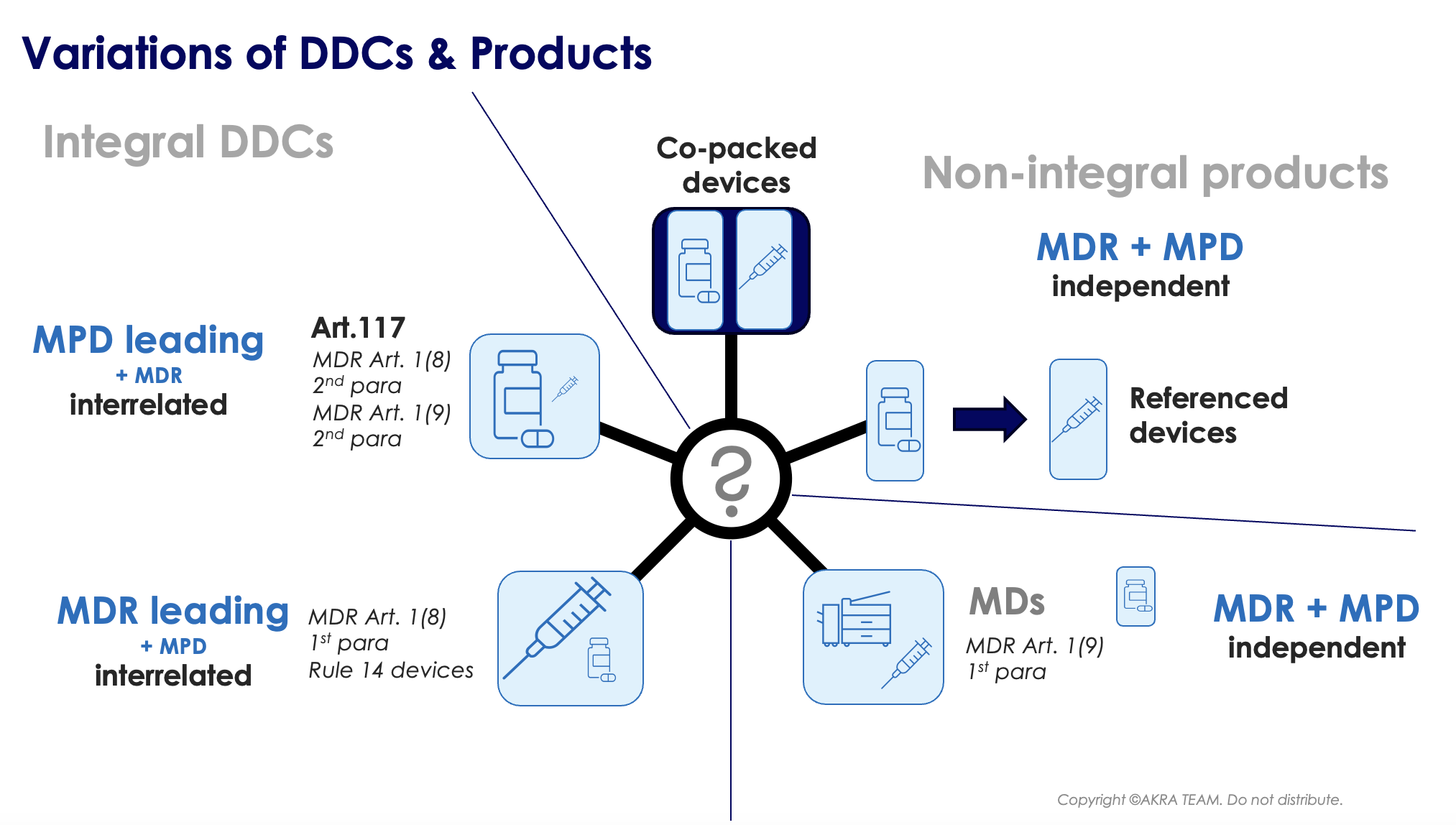
Integral DDC products: These comprise mainly the following 2 product groups:
- Integral DDCs, in which the Medical Device (MD) has the primary mode of action
- Integral DDCs, in which the pharmaceutical component (PC) has the primary mode of action
Integral DDCs / MD primary mode of action
These devices relate to article 1(8) 2nd paragraph. This product group comprises devices that are MDs, incorporating as an integral part a medicinal product. The principal mode of action is achieved by the MD part. The Medicinal Product has an ancillary mode of action to that of the MD.
Processes involved for marketing of these devices are a conformity assessment with a designated notified body (NB) for the MD part. The focus of the NB regarding the PC part is the assessment of usefulness of the included PC to the device. Finally, a consultation of a designated medicinal CA/EMA by the notified body on the safety, quality, and usefulness of the included PC will be initiated (EMA will be involved, if PC is derived from blood or plasma).
Examples for such DDC products acc. to MDCG 2022-5 are displayed in the following.
Examples: Devices containing an integral ancillary medicinal substance
- Catheters coated with heparin or an antibiotic agent
- Bone cements containing antibiotic
- Root canal fillers which incorporate medicinal products with ancillary action to that of the device
- Soft tissue fillers incorporating local anaesthetics
- Bone void filler containing growth factors
- Condoms coated with spermicides
- Electrodes with steroid-coated tip
- Wound dressings, surgical or barrier drapes (including tulle dressings) with antimicrobial agent
- Intrauterine contraceptives containing copper or silver
- Ophthalmic irrigation solutions principally intended for irrigation which contain components that support the metabolism of the endothelial cells of the cornea
- Drug eluting coronary stents
- Blood bags containing substances which, if used separately, would be considered to be a medicinal product
- Liquid wound dressing with antimicrobial agent
Source: MDCG 2022-5
Integral DDCs / PC primary mode of action
Products, falling into this category, are products acc. to article 1(8) 2nd paragraph of the EU-MDR. I.e. Devices are MDs, incorporating as an integral part a PC, whereas the principal mode of action is achieved by the PC.
In addition, products acc. to article 1(9) 2nd paragraph of the EU-MDR. I.e. devices intended to administer a medicinal product, which (a) form a single integral product with a PC, which is (b) intended exclusively for use in the given combination and (c) which is not reusable.
The following list shows examples for these group of products acc. to MDCG 2022-05.
Examples for integral DDCs
- Syringes prefilled with a medicinal product
- Aerosols containing a medicinal product
- Nebulisers pre-charged with a specific medicinal product
- Patches for transdermal drug delivery
- Implants containing medicinal products in a polymer matrix whose purpose is to release the medicinal product, for example plastic beads containing antibiotic for treating bone infections, or a matrix to release osteoinductive proteins into the surrounding bone
- Intrauterine contraceptives whose purpose is to release progestogens
- Single-use disposable iontophoresis devices incorporating a medicinal product, whose purpose is to deliver the medicinal product for the treatment of a medical condition
- Wound treatment products comprising a matrix whose purpose is the administration of medicinal products, for example wound dressings containing an antimicrobial agent where the primary action of the dressing is to administer the agent to the wound for the purpose of controlling infection
- Temporary root canal fillers incorporating medicinal products, whose purpose is to deliver the medicinal product
- Tablets containing a medicinal product with embedded sensor to monitor adherence to treatment
Source: MDCG 2022-5
A novelty with the EU-MDR is article 117. Article 117 represents an amendment to the Directive 2001/83/EC in such a way that pharmaceutical manufacturers must provide evidence to the competent authority, when submitting the marketing authorization dossier, that the MD part of the DDC complies with the applicable GSPRs as set out in Annex I to the EU-MDR.
Thus, the following options apply:
- The manufacturer provides an EU declaration of conformity, or
- The relevant certificate issued by a notified body allowing the manufacturer to affix a CE marking to the medical device, or
- The submission of a notified body opinion
The latter route is the less burdensome for pharmaceutical manufacturers, provided that the MD part does not bear a CE-Mark. However, cooperation with notified bodies may be new to a pharmaceutical manufacturer and is different from working with competent authorities.
The resulting challenges range from “How to find a notified body”, via how to cooperate with a NB (incl. timelines) up to the questions, what documents to be submitted and in which format.
AKRA TEAM can help you in all these stages to facilitate the way through the processes.
Support & Training
Contact AKRA TEAM for support, hands on implementation services and personalized training by experts with key competencies in the areas listed below.
Key points
DDCs represent an emerging sector in healthcare industry, comprising a heterogeneous group of different products, which combine a medical device with a medicinal product.
Thus, both legislations, the EU-MDR and the Medicinal Products Directive are applicable, whereas the leading legislation is determined by the principal mode of action of the product.
Specifically, Article 117 of the EU-MDR leads to significant changes to the regulatory landscape of various stakeholders and requires adoption to already established processes. Compliance with the applicable GSPRs acc. to Annex I EU-MDR must be demonstrated when submitting a marketing authorization dossier for DDCs, for which the primary mode of action is based on the pharmaceutical. In contrary to the past, a notified body must be involved, provided that the specific device part does not bear a CE-Mark supported by a declaration of conformity and/or a certificate from a designated notified body.
AKRA TEAM can support you working thru this process effectively and efficiently.
Our Services
Selecting a notified body, designated for the respective MD part.
Communication support with the NB.
Consultancy prior to submitting documents to the NB.
Preparation of documentation fulfilling Article 117 obligations.
Gap-Assessment / readiness check of documents including detailed recommendations towards compliance
Consultancy on Non-Conformities / Deficiencies raised by the notified body. Support on response letters to the notified body.
Webinar/Training on Drug-Device-Combinations (Overview / Processes)