Risk management according to the European legislation and EN ISO 14971:2019
Clinical Performance Studies under the IVDR – What’s required, what has changed?
General Background
According to Annex I, Section 3 of Regulation (EU) 2017/45 (EU MDR) manufacturers of medical devices are required to establish, implement, document, and maintain a risk management system to receive approval for their devices in Europe. The harmonized standard EN ISO 14971:2019 defines the specific risk management activities in more detail than provided by the MDR. ISO 14971 provides manufacturers with a framework for application of a systematic approach to manage the risks associated with the use of their medical devices – including in vitro diagnostic medical devices – and is also formally recognized as the risk management standard by regulatory authorities outside of the EU, including the US, Canada, and Australia.
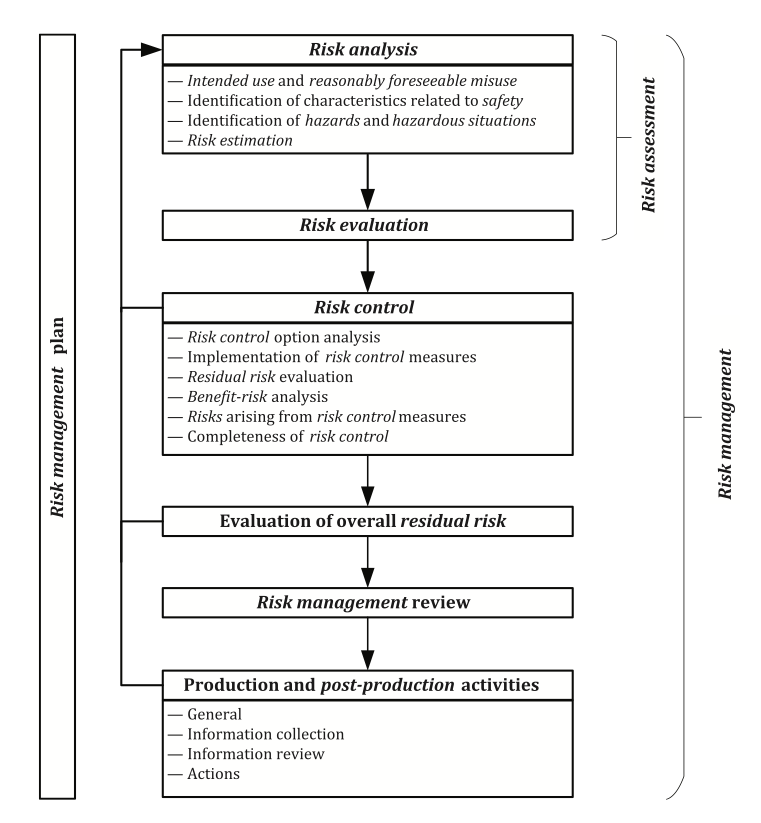
ISO 14971 illustrates the requirements for a risk management process as part of the manufacturers risk management system in a helpful schematic representation:
Key considerations for manufacturers under the EU IVDR include:
According to this figure it is evident that all risk management activities must be planned. The risk management plan documents a roadmap for all risk management activities conducted throughout the entire life cycle of the medical device. While considering the intended purpose of the medical device the risk management plan defines the criteria for risk acceptability which will ensure an objective evaluation of the residual risks later in the process.
Risk assessment consists of the risk analysis and risk evaluation. A prerequisite for a risk analysis is the clear description of the intended use allowing a discrimination between the correct use or correct application of the medical device and its misuse. The manufacturer needs to define and document the reasonably foreseeable misuse and consider it in the risk management process. Reasonably foreseeable misuse includes the intentional use of the medical device for an application that is unspecified or unintended by the manufacturer or an unintentionally performed use error.
Risk analysis further requires identifying the characteristics of the medical device that can affect its safety which may be related to the performance or the operating principle of the medical device. The technical report ISO/TR 24971 provides guidance on the application of ISO 14971 and includes in Annex A an extensive list of questions that can assist the manufacturer in identifying the characteristics related to safety. The safety characteristics can be qualitative or quantitative and it may be necessary to establish certain limits that should be adhered to.
The next step is the identification of the hazards associated with the medical device and identifying the reasonably foreseeable sequences or combinations of events that can lead to hazardous situations. Different sequences of events can lead from one hazard to different hazardous situations, and one specific hazardous situation can lead to diverse kinds and severities of harm according to the individual circumstances with each situation representing a separate risk.
Risk estimation for each of the identified hazardous situations requires an estimation of the severity of any possible harm and of the probability that this harm occurs. Various tools exist for the risk analysis and the manufacturer may decide which is most appropriate (e.g., Preliminary Hazard Analysis, Fault Tree Analysis, Event Tree Analysis, Failure Mode and Effects Analysis, Hazard and Operability Study, Hazard Analysis and Critical Control Point).
Risk acceptance will be estimated according to the risk acceptability matrix defined in the risk management plan which comprises the risk evaluation. It is important to note that all hazardous situations and all kinds of harm need to be considered. Focusing only on the worst-case scenarios with the highest severity of harm would ignore scenarios with less severe harm which may be more probable to occur and could potentially lead to a higher risk. The conclusions of the risk evaluation must be documented in the risk management file. Annex C.6 of ISO/TR 24971 provides examples how to determine acceptability of risks considering several elements and approaches for risk evaluation: regulatory requirements, international standards, state of the art, and stakeholder concerns. Any risk judged as acceptable directly becomes the residual risk. For any risk not judged acceptable it is mandatory to implement risk control measures.
There are several risk control options for eliminating or reducing risks to an acceptable level. They must be performed in the following order:
- eliminating the risk by making the design of the medical device and its manufacturing process inherently safe
- implementing protective measures in the design of the medical device or in the manufacturing process, thereby
- reducing the probability of occurrence of a hazardous situation or harm and/or
- reducing the severity of the harm
- providing information for safety to the users of the medical device (i.e., warnings or contraindications, or instructions on how to handle and use the medical device)
Designated risk control measures must be implemented, and the implementation as well as the effectiveness of the implemented risk control measures must be verified, and the risk management file must document the results of these verifications.
Any residual risk must be estimated and evaluated again after implementation of the risk control measures according to the predefined risk acceptability criteria. For any risk not judged acceptable, it is necessary to consider further risk control measures according to an iterative process between risk control and risk assessment. If the manufacturer concludes after thorough analysis that further risk reduction is not possible, a benefit–risk analysis may be performed.
Benefit-risk analysis
For a comparison of benefits and risks several aspects are to be considered according to ISO/TR 24971:
- characterization of the disease or condition of the intended patients
- the uncertainty of data (An initial literature search for the hazards and the medical device being considered can provide insight into the balance between benefit and risk)
- production and post-production information for similar medical devices that are already available on the market
- the generally acknowledged state of the art
- a comparison of the benefits of the medical device under development with the benefits of similar medical devices available on the market
- a comparison of the residual risks of the medical device under development with the residual risks of similar medical devices available on the market.
Such a collection and analysis of data and literature may allow to determine if the benefits of using the medical device outweigh the residual risk. In case there is no such conclusion, the manufacturer needs to consider a modification of the medical device eliminating the specific risk. Alternatively, a restriction of the intended use may be required, which, e.g., may be the exclusion of a vulnerable patient population.
The manufacturer is required to check that all identified hazardous situations have been addressed and all risk control activities have been completed. In case the selected and implemented risk control measures introduce new risks, those need to be analyzed, evaluated, and controlled as well.
According to ISO 14971 it is required that the contributions of all individual residual risks are considered, and that the overall residual risk is evaluated in relation to the benefits of the intended use of the medical device.
ISO/TR 24971 provides more detailed guidance on possible approaches that can be used in the evaluation and on inputs and other aspects that can be considered. It is described that the criteria for acceptability of the overall residual risk can be different from the criteria for acceptability of individual risks. The guidance emphasizes that the criteria used to evaluate the overall residual risk are often based on additional elements, such as the benefits of the intended use of the medical device. The manufacturer is responsible for determining an appropriate method for evaluating the overall residual risk and there is no preferred way how to do it.
Some examples of approaches to evaluate the overall residual risk are:
- weighing the benefits related to the intended use of the medical device against the overall residual risk – considering that benefits can be described by their magnitude or extent, the probability of experiencing the benefit within the intended patient population, and the duration and frequency of the benefit, and taking into account the knowledge of the intended medical indication, the generally acknowledged state of the art in technology and medicine, and the availability of alternative medical devices or treatment options
- considering visual representations of the residual risks
- comparing the medical device under consideration to similar medical devices available on the market, including current information on intended use and adverse events of similar medical devices, as well as information from scientific literature, including information about clinical experience
- considering use of expert judgment to support the evaluation of the overall residual risk in relation to the benefits expected from using the medical device
- consider further investigation of some risks (many risks may be close to being not acceptable and the overall residual risk might not be deemed acceptable without further investigation)
- further investigation may also be required when some risks are interdependent with respect to either their causes or the risk control measures applied
To receive approval for a device the manufacturer must conclude that the medical device under consideration has a favorable benefit–risk ratio.
The manufacturer is also required to inform users of any significant residual risks and to disclose those risks by providing relevant information in the accompanying documentation of the medical device. The disclosure of residual risks is distinct from information for safety, representing a risk control measure. It must be acknowledged that information for safety is instructive and provides the user with information, e.g., on how to use the medical device, on any actions to take, or on any actions to avoid preventing a particular hazardous situation or harm from arising.
After the design and development of the medical device and before its commercial distribution, the manufacturer is required to review that the risk management plan was properly executed and appropriately implemented. The risk management review is also intended to ensure that the overall residual risk is acceptable, and that appropriate methods are in place to collect and review relevant production and post-production information. The risk management report is created after the review of the execution of the risk management plan. The manufacturer needs to ensure that all information provided is aligned and consistent across the technical documentation.
The risk management file created by the manufacturer needs to be updated according to information from the production phase and from the post-market surveillance system. This relates to a continuous evaluation of hazards and their frequency of occurrence, estimates of their associated risks, as well as on the overall risk, benefit-risk ratio, and risk acceptability and will allow a conclusion on the necessity to amend or implement additional control measures.
The clause on production and post-production information in ISO 14971 precisely elaborates on the requirements and activities in four sections corresponding to the separate steps that the manufacturer needs to ensure are implemented in the risk management system (additional examples are provided in ISO/TR 24971):
- Establishing a system to collect and review relevant production and post-production information and define the necessary activities in the risk management plan; including the definition of appropriate methods for the collection and processing of data, such as statistical methods for trend analysis; it is suggested to integrate monitoring and feedback processes required by a quality management system
- Proactively collecting relevant information for the medical device under consideration (such as information from users, from the supply chain and on the generally acknowledged state of the art (such as new or revised standards, alternative medical devices or alternative therapies), publicly available information about similar medical devices and similar other products on the market
- Review of the collected information is relevant to the safety of the medical device (determination whether a previously unidentified hazard or hazardous situation exists, an estimated risk is no longer acceptable, the benefits of the medical device no longer outweigh the overall residual risk, or the generally acknowledged state of the art has changed)
- Initiating necessary actions in case of identification of relevant safety information (including a review the risk management file and determination if any new risk needs to be assessed or any previously estimated risk needs to be assessed again, and if it is necessary to implement additional risk control measures; estimation if actions regarding medical devices already on the market are required; evaluating the impact on risk management activities previously performed potentially providing valuable input for top management when they review the suitability of the risk management process)
Support & Training
Contact AKRA TEAM for support, hands on implementation services and personalized training by experts with key competencies in the areas listed below.
Key points
Key changes that require the manufacturer´s immediate attention:
Compliance with the EU MDR requires the manufacturer to establish, implement, document, and maintain a risk management system.
Specific risk management activities are defined in the harmonized standard EN ISO 14971:2019 and additional guidance on the application of the requirements of the standard is laid out in the technical report ISO/TR 24971.
A clear description of the intended use is a prerequisite for the risk analysis allowing a discrimination between the correct use or correct application of the medical device and its misuse. The intended use includes the medical indication and application (disease type, tissue and part of the body), the intended target treatment population (children, adults, elderly or specific patient groups such as pregnant and lactating individuals), the users and the use environment (lay users at home, professional users in or outside a clinical environment), as well as the operating principle (how the diagnosis or treatment is achieved).
The risk management plan is expected to describe a specification of methods that are used to examine qualitative and quantitative aspects of clinical safety with clear explanation how residual risks and side-effects are determined.
The overall residual risk of a medical device must be acceptable, and the manufacturer must be able to provide adequate justifications that the benefit-risk ratio for the medical device is favorable.
Our Services
Even though the harmonized standard provides a framework for the risk management process there are still many challenges when implementing the individual process steps for a specific medical device. AKRA TEAM can help you fulfilling your legal obligations in a practical and reasonable way by mitigating your risk and increasing your certification success.
Training
AKRA TEAM can provide training on implementing and maintaining a risk management system according to the requirements laid out in the EU MDR and the harmonized standard EN ISO 14971:2019. This will ensure that your personnel have the appropriate training to perform the risk management tasks assigned to them.
Process and Templates Development
AKRA TEAM can assist creating required SOPs for your risk management process including defining post-production activities as well as providing templates for risk management plan and risk management report.
Gap Assessment
AKRA TEAM offers reviewing your risk management file and providing a gap assessment on all parts of the file, especially, the risk management process, the risk management plan and report, as well as the risk analysis including risk estimation and risk control measures, the conclusion of the benefit-risk evaluation and completeness of disclosed residual risks.
Implementation
AKRA TEAM can support you in the implementation of your defined risk management activities ensuring that the risk management plan is properly executed and confirming that all information provided is aligned and consistent across the technical documentation. This will also ensure that all clinical risks identified in the clinical evaluation are covered in the risk file.
Continuous update of documentation
AKRA TEAM can help updating your documentation according to information from the production phase and from the post-market surveillance system. This may include improving your processes to facilitate continuous update and evaluation of hazards and their frequency of occurrence, estimates of their associated risks, as well as on the overall risk, benefit-risk ratio, and risk acceptability.
Interested in our services?
Lorem ipsum dolor sit amet, consec tetur adipis cing elit. Ut elit tellus, luctus nec ullam corper mattis, pulvinar dapibus leo.