Clinical investigations under EU MDR
What’s required for the most suitable clinical investigation strategy?
General Background
Clinical investigations under the EU Medical Device Regulation (EU MDR) refer to the process of conducting clinical investigations to collect data on the safety and performance of medical devices before they are placed on the market. This may also include significant changes and initial CE marking under EU MDR where clinical investigation data is deemed necessary to address gaps in clinical evidence. These investigations are an important part of the EU regulatory process and are required for certain classes of medical devices to ensure that they meet relevant safety and performance standards.
With the EU MDR clinical investigations are more important than ever. Especially since conformity assessment based on equivalence is often no longer possible, medical device manufacturers must collect clinical data on their own device(s) in order to achieve successful conformity assessment.
For class III devices and implantable devices data should generally be sourced from clinical investigations that have been carried out under the responsibility of a sponsor. This also applies to lower class devices that have a new intended use or incorporate new technologies that raise safety or other concerns. EU MDR requires that clinical investigations be conducted for devices that raise safety concerns or have a high degree of innovation, regardless of their classification.
Before conducting a clinical investigation, the sponsor of the study must obtain approval from the relevant Competent Authority in the EU member state where the investigation is to take place. The sponsor must also obtain ethical approval from an Ethics Committee, which is responsible for reviewing the study protocol to ensure that it is ethically sound and that the rights and safety of study participants are protected.
Clinical investigations under the EU MDR must be conducted in accordance with Good Clinical Practice (GCP) guidelines, which provide a set of international standards for the design, conduct, and reporting of clinical trials. The guidelines aim to ensure that clinical trials are conducted in a way that protects the rights, safety, and well-being of study participants, and that the data generated is reliable and can be used to support regulatory decision-making.
Supporting Subheading
An Attractive Call to Action Text
Once a clinical investigation has been completed, the sponsor must prepare a clinical investigation report (CIR), which summarizes the results of the study and provides conclusions on the safety and performance of the medical device. The CIR is then submitted to the Competent Authority for review as part of the conformity assessment process.
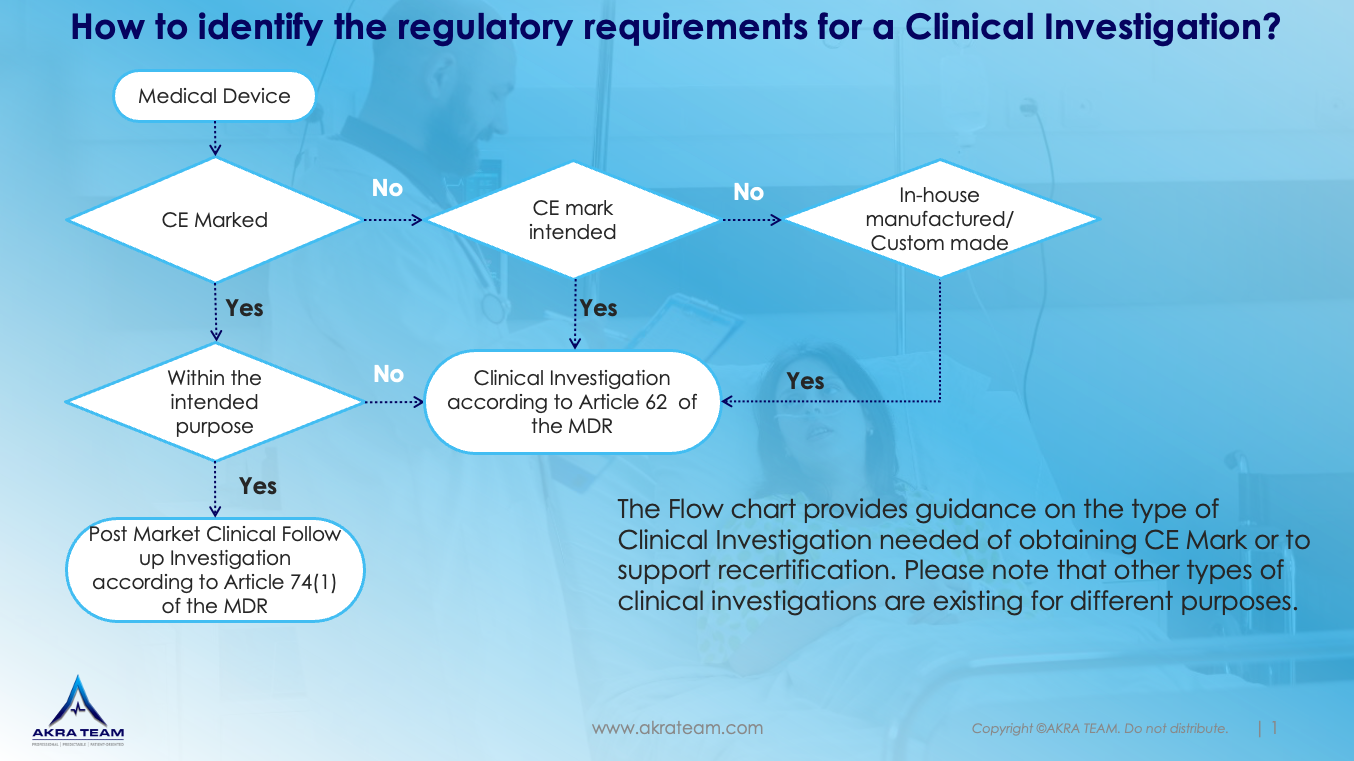
Planning and conducting a clinical investigation accordingly are often resource intensive and dependent on the applicable type of Clinical investigations different requirements have to be addressed (See flowchart). Akra Team can support you in your effort to design and plan a clinical investigation reasonably.
Support & Training
Contact AKRA TEAM for support, hands on implementation services and personalized training by experts with key competencies in the areas listed below.
Key considerations
Key changes that require the manufacturer´s immediate attention:
Align your clinical investigation strategy with your marketing strategy: What are the product claims you plan to make and which data support them?
Generate enough data to get your stakeholders trust in the safety and performance of your device.
Make sure to consider your risk management data when planning your clinical investigation outcome parameters.
Consider reimbursement requirements within your market and regulatory requirements of other potential target markets.
Confirm that your QMS system is covering your clinical investigation, even if some activities are outsourced.
Apply country specific requirements for clinical investigation application and documentation.
Our Services
Training
AKRA TEAM can train you in the general regulatory requirements for clinical investigations of the MDR and the corresponding guidance documents endorsed by the Medical Device Coordination Group (MDCG).
AKRA TEAM can provide training on ISO 14155 “Clinical investigation of medical devices in humans – Good clinical practice"
Process and Templates Development
AKRA TEAM can aid in the creation of a clinical investigation procedure, related QMS procedures (e.g. Clinical Evaluation Plan) as well as relevant templates.
Gap Assessment / Guidance
AKRA TEAM can perform a GAP analysis of your existing data and/ or your Clinical Investigation plan to identify missing information and to provide guidance on the best approach to close the gaps.
AKRA TEAM can help your company with the identification of the most suitable clinical strategy for your product.
AKRA TEAM ensures that the design of your clinical investigation is in line with well-established international guidance documents such as the international standard ISO 14155 “Clinical investigation of medical devices in humans – Good clinical practice” as well as the ethical principles set out in the latest version of the “Declaration of Helsinki” of the World Medical Association as required per MDR article 1 (64).
Implementation
AKRA TEAM can provide feedback in clinical investigation strategy, protocol development, CRF design, informed consent design. Support can also be provided with trial submission at Competent Authorities and Ethics Committees as well as site initiation, study start-up, and close-out activities.
Continuous update and documentation
Once EU MDR compliant documentation has been implemented and submitted to the Notified Body, it may require continuous update due to requirements such as for PMS or due to any other changes triggered by the regulators, the market, the data on the device or by the manufacturer itself. Akra team will ensure follow-up and can accompany manufacturers to maintain compliance.
Interested in our services?
Lorem ipsum dolor sit amet, consec tetur adipis cing elit. Ut elit tellus, luctus nec ullam corper mattis, pulvinar dapibus leo.